物理理论改进了蛋白质折叠预测
蛋白质是执行生命必需的多种功能的重要分子。为了正常发挥作用,许多蛋白质必须折叠成特定的结构。然而,蛋白质折叠成特定结构的方式仍然很大程度上未知。东京大学的研究人员开发了一种新颖的物理理论,可以准确预测蛋白质如何折叠。他们的模型可以预测以前模型无法预测的事情。提高蛋白质折叠知识可以为医学研究以及各种工业过程带来巨大好处。
该研究题为“通过基于简单结构的统计力学模型准确预测蛋白质折叠机制”,发表在《自然通讯》上。
你实际上是由蛋白质组成的。这些链状分子由数十到数千个称为氨基酸的小分子组成,形成头发、骨骼、肌肉、消化酶、对抗疾病的抗体等。蛋白质通过折叠成各种结构来制造这些东西,这些结构反过来又形成这些更大的组织和生物成分。
通过更多地了解这种折叠过程,研究人员可以更好地了解构成生命本身的过程。这些知识对于医学也至关重要,不仅对于开发新的治疗方法和生产药物的工业流程,而且对于了解某些疾病如何发生,因为有些疾病是蛋白质折叠出错的例子。因此,说蛋白质很重要是温和的说法。蛋白质是生命的物质。
受到蛋白质折叠重要性的鼓舞,艺术与科学学院的项目助理教授KojiOoka和生命科学系和物理系的MunehitoArai教授开始着手改进蛋白质折叠的预测方法。由于多种原因,这项任务十分艰巨。特别是,模拟分子动力学的计算要求需要强大的超级计算机。
最近,基于人工智能的程序AlphaFold2准确预测了给定氨基酸序列产生的结构;但它无法提供蛋白质折叠方式的细节,使其成为一个黑匣子。这是有问题的,因为蛋白质的形式和行为各不相同,以至于两个相似的蛋白质可能以完全不同的方式折叠。因此,研究人员需要一种不同的方法,而不是人工智能:统计力学,物理理论的一个分支。
WSME的四次迭代(从原始版本到新版本)以及两个针对更具体情况的专用版本。图片来源:©2023Ooka&AraiCC-BY
“20多年来,一种名为Wako-Saitô-Muñoz-Eaton(WSME)模型的理论已经根据天然蛋白质结构成功预测了包含约100个或更少氨基酸的蛋白质的折叠过程,”Arai说。
“WSME一次只能评估蛋白质的一小部分,错过了相距较远的部分之间的潜在连接。为了克服这个问题,我们创建了一个新模型WSME-L,其中L代表“连接子”。我们的接头对应于这些非局部相互作用,并允许WSME-L阐明折叠过程,而不受蛋白质大小和形状的限制,而AlphaFold2则无法做到这一点,”Arai补充道。
但事情并没有就此结束。Ooka和Arai着眼于现有蛋白质折叠模型的其他局限性。蛋白质可以存在于活细胞内部或外部;内部的那些在某种程度上受到细胞的保护,但是细胞外部的那些,例如抗体,在折叠过程中需要额外的键,称为二硫键,这有助于稳定它们。传统模型无法考虑这些键,但WSME-L的扩展(称为WSME-L(SS))可以考虑这些键,其中每个S代表硫化物。
更复杂的是,一些蛋白质在折叠开始之前就具有二硫键,因此研究人员进行了进一步的增强,称为WSME-L(SS完整),它以额外的计算时间为代价来考虑这种情况。
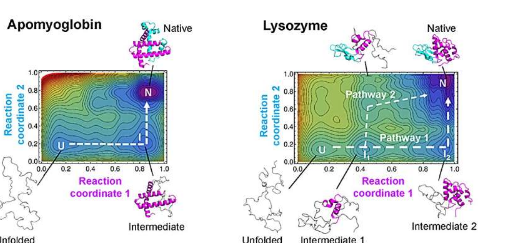
“我们的理论使我们能够在相对较短的时间内绘制出一种蛋白质折叠途径图;假设天然蛋白质结构可通过实验或AlphaFold2预测,”Arai说。
“由此产生的景观可以全面了解长蛋白质可能采取的多种潜在折叠途径。最重要的是,我们可以仔细检查瞬态结构。这可能对研究阿尔茨海默氏症和帕金森氏症等疾病有帮助——这两种疾病都是由蛋白质折叠失败引起的。正确折叠。此外,我们的方法可能有助于设计新型蛋白质和酶,这些蛋白质和酶可以有效折叠成稳定的功能结构,用于医疗和工业用途,”Arai继续说道。
虽然这里产生的模型准确地反映了实验观察结果,但Ooka和Arai希望它们可以用来阐明许多尚未进行实验研究的蛋白质的折叠过程。人类大约有20,000种不同的蛋白质,但仅对其中的大约100种蛋白质的折叠过程进行了彻底研究。
免责声明:本文由用户上传,与本网站立场无关。财经信息仅供读者参考,并不构成投资建议。投资者据此操作,风险自担。 如有侵权请联系删除!
-
【木地板材料】木地板是一种广泛应用于家居和商业空间的地面装饰材料,因其自然美观、耐用性强、环保性好等特...浏览全文>>
-
【木地板安装】木地板安装是一项需要细致规划和专业操作的工程,无论是新房装修还是旧房翻新,选择合适的木地...浏览全文>>
-
【木的组词怎么写】在汉语学习中,词语的积累和运用是非常重要的部分。对于“木”这个字来说,它不仅是常见的...浏览全文>>
-
【木的象形字有哪些字】在汉字的发展过程中,许多字最初都是通过象形的方式创造出来的,用来表示具体的事物。...浏览全文>>
-
【木岛法子介绍】木岛法子(Kazuko Kikuchi)是日本著名演员、模特及艺人,以其在影视作品中的出色表现和独特...浏览全文>>
-
【木代尔是什么面料】“木代尔是什么面料”是许多消费者在选购衣物时常常会提出的问题。木代尔是一种天然纤维...浏览全文>>
-
【木代尔和莫代尔哪种面料好】在选择衣物面料时,很多人会遇到“木代尔”和“莫代尔”这两个名称,容易混淆它...浏览全文>>
-
【萝卜的营养价值】萝卜是一种常见的根茎类蔬菜,不仅味道清脆爽口,还具有丰富的营养价值。无论是生吃、炒食...浏览全文>>
-
【萝卜的药用功效和作用】萝卜,作为日常生活中常见的蔬菜之一,不仅味道清脆、营养丰富,还具有多种药用价值...浏览全文>>
-
【萝卜的家常做法】萝卜是一种非常常见的蔬菜,不仅价格实惠,而且营养丰富,适合多种烹饪方式。无论是炖、炒...浏览全文>>